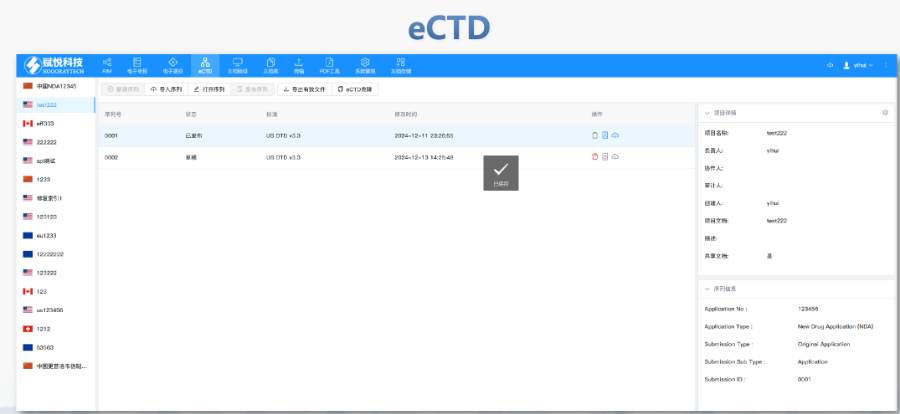
商機詳情 -
太倉原料藥eCTD使用
FDA圍繞eCTD發(fā)布了10余項法規(guī)指南,涵蓋格式要求、文件生命周期、數(shù)據(jù)安全等細(xì)節(jié),其中《ICH M2 EWG》作為綜合性技術(shù)文件,成為企業(yè)申報的參考。eCTD的實施提升了審評效率,通過標(biāo)準(zhǔn)化XML結(jié)構(gòu)和電子簽章技術(shù),減少了紙質(zhì)遞交的物流與時間成本,同時支持全生命周期管理,便于后續(xù)變更和補充資料的動態(tài)更。 美國在eCTD實施中注重與ICH國際標(biāo)準(zhǔn)的兼容性,例如采用統(tǒng)一的CTD模塊化結(jié)構(gòu)和PDF技術(shù)規(guī)范。然而,其區(qū)域性要求(如信封信息中的Application ID、Submission Subtype)仍體現(xiàn)本土化特色。這種“國際框架+本地適配”的模式,既保障了跨國藥企的申報便利,又滿足了FDA的監(jiān)管需求。澳大利亞NDA注冊申報相關(guān)技術(shù)支持。太倉原料藥eCTD使用
DMF維護(hù)與合規(guī) ?年度更 即使無變更,每年需提交聲明;重大工藝/設(shè)施變更需及時通知客戶并更文件。 ?現(xiàn)場檢查 原料藥企業(yè)需通過FDA現(xiàn)場檢查,驗證是否符合ICH Q7 GMP標(biāo)準(zhǔn),并與DMF內(nèi)容一致。 ?轉(zhuǎn)讓與關(guān)閉 ?轉(zhuǎn)讓:需書面通知FDA并提供持有者信息。 ?關(guān)閉:未提交年度報告或持有人主動申請,需說明原因并通知所有授權(quán)方。 關(guān)鍵注意事項 ?數(shù)據(jù)質(zhì)量:所有資料需準(zhǔn)確、完整,減少審核延遲風(fēng)險。 ?合規(guī)性:遵循FDA指南(如21 CFR Part 207)及USP標(biāo)準(zhǔn)(如培養(yǎng)基物料來源級別)。 ?溝通機制:建議通過專業(yè)機構(gòu)(如瑞歐佰藥)協(xié)助,定期提交周報并制定計劃表以提高效率。 常見問題解答 ?生物制品分類:培養(yǎng)基、外泌體等均屬Ⅱ類DMF。 ?質(zhì)量標(biāo)準(zhǔn):參考USP及同行標(biāo)準(zhǔn),需提供分析方法驗證及雜質(zhì)對比研究。 ?周期估算:資料準(zhǔn)備約5-50個工作日,總周期受缺陷回復(fù)影響。蕪湖NDAeCTD哪個品牌好美國ANDA注冊申報相關(guān)技術(shù)支持。
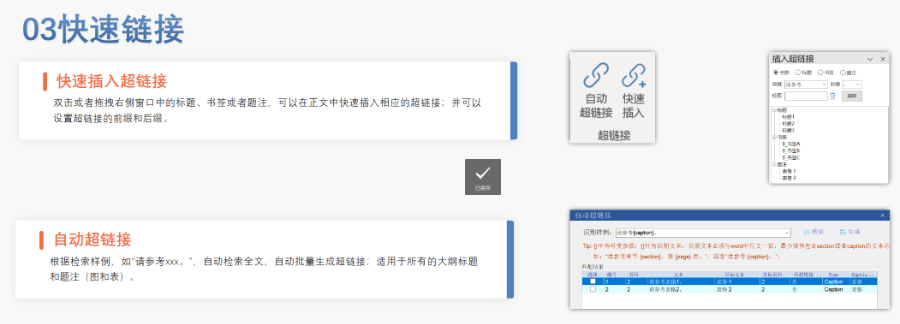
多國審評程序與eCTD遞交途徑的適配:歐盟藥品審評程序包括集中(CP)、分散(DCP)、互認(rèn)(MRP)和國家程序(NP),eCTD需適配不同程序的遞交要求。例如: ?集中審評程序(CP)?:通過EMA的eSubmission Gateway提交,審評時限約240個工作日,eCTD需包含完整的模塊1-5及多語言標(biāo)簽文件。 ?分散審評程序(DCP)?:需通過CESP(歐盟共同提交門戶)遞交,參考成員國(RMS)主導(dǎo)審評,eCTD需支持多國同步評估的模塊化拆分。 ?互認(rèn)程序(MRP)?:已授權(quán)成員國作為RMS,eCTD需包含基線序列(Baseline Sequence 0000)以整合歷史審評數(shù)據(jù),并通過CMDh協(xié)調(diào)分歧。
危機應(yīng)對與應(yīng)急遞交機制 在公共衛(wèi)生緊急事件(如COVID-19)中,EMA允許簡化eCTD序列,優(yōu)先審評關(guān)鍵模塊并暫緩非數(shù)據(jù)。申請人可通過快速通道(Fast Track)提交疫苗或藥物的eCTD資料,審評周期可壓縮至6個月。此類申請需附風(fēng)險評估報告,并承諾后續(xù)補交完整數(shù)據(jù)。 數(shù)據(jù)安全與長期存檔 歐盟要求eCTD資料存檔期限至少為藥品上市后30年,EMA采用分布式存儲和區(qū)塊鏈技術(shù)確保數(shù)據(jù)不可篡改。申請人需定期備份本地副本,并使用符合GDPR要求的加密傳輸協(xié)議(如AS2)遞交。歷史數(shù)據(jù)的遷移和格式轉(zhuǎn)換(如NeeS轉(zhuǎn)eCTD)需遵循特定技術(shù)規(guī)范。 環(huán)保效益與可持續(xù)發(fā)展 eCTD取代紙質(zhì)遞交后,歐盟每年減少約500噸紙張消耗,審評流程的數(shù)字化降低碳足跡約30%。虛擬審評會議和電子簽名進(jìn)一步減少了差旅需求,契合歐盟2050碳中和目標(biāo)。未來,eCTD4.0將通過數(shù)據(jù)壓縮技術(shù)進(jìn)一步降低服務(wù)器能耗。歐盟DMF注冊申報相關(guān)技術(shù)支持。
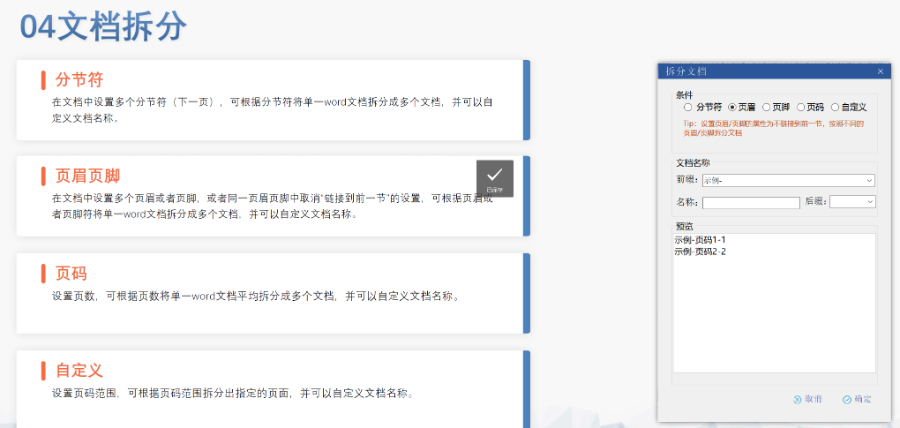
電子遞交的合規(guī)性與風(fēng)險管理 歐盟要求申請人確保電子資料與紙質(zhì)版本完全一致,若未在規(guī)定時間提交紙質(zhì)文件可能導(dǎo)致注冊終止。驗證過程中,“錯誤”級別問題(如文件命名不規(guī)范、XML邏輯錯誤)必須修正,而“警告”和“提示信息”則建議優(yōu)化以提升審評體驗。EDQM和EMA均提供驗證工具,申請人需在遞交前完成內(nèi)部預(yù)驗證。 官方費用結(jié)構(gòu)與支付流程 歐盟eCTD遞交費用因?qū)徳u程序類型而異:集中程序費用較高,涵蓋科學(xué)評估和合規(guī)審查成本;國家程序費用由各成員國自行設(shè)定。CEP申請需向EDQM支付評審費,具體金額根據(jù)原料藥類型和變更復(fù)雜度分級。繳費需通過官方指定渠道完成,并附上付款憑證作為模塊1的組成部分。 多語言支持與翻譯要求 盡管歐盟允許使用英語提交,但部分成員國要求模塊一的行政文件翻譯為本地語言。臨床試驗數(shù)據(jù)庫(如SDTM和ADaM)需以英語呈現(xiàn),同時提供雙語標(biāo)簽以支持多國審閱。專業(yè)翻譯服務(wù)在確保技術(shù)術(shù)語準(zhǔn)確性方面至關(guān)重要,尤其針對復(fù)雜藥學(xué)和非臨床數(shù)據(jù)。eCTD驗證標(biāo)準(zhǔn)相關(guān)技術(shù)支持。工業(yè)園區(qū)NDAeCTD哪個品牌好
歐盟CESP提交通道相關(guān)技術(shù)支持。太倉原料藥eCTD使用
從紙質(zhì)到電子的歷史過渡 2017年前,美國允許紙質(zhì)與eCTD并行提交,但此后逐步淘汰紙質(zhì)通道,保留緊急情況下的例外審批。2020年電子化后,所有IND、NDA、ANDA和DMF強制采用eCTD格式。 ?系統(tǒng)平臺升級 FDA通過“藥品業(yè)務(wù)應(yīng)用系統(tǒng)”和“藥品eCTD注冊系統(tǒng)”實現(xiàn)電子資料接收、受理與審評的全流程數(shù)字化。2022年系統(tǒng)增自動推送受理文書和短信提醒功能,減少人工干預(yù)。 ?電子文檔結(jié)構(gòu)優(yōu)化 美國eCTD采用分層文件夾結(jié)構(gòu),例如化學(xué)藥品的模塊1-5分別對應(yīng)行政文件、總結(jié)報告、質(zhì)量數(shù)據(jù)等。2020年后增“臨床試驗數(shù)據(jù)庫”裝盒要求,強化數(shù)據(jù)可追溯性。太倉原料藥eCTD使用
賦悅科技(杭州)有限責(zé)任公司在同行業(yè)領(lǐng)域中,一直處在一個不斷銳意進(jìn)取,不斷制造創(chuàng)新的市場高度,多年以來致力于發(fā)展富有創(chuàng)新價值理念的產(chǎn)品標(biāo)準(zhǔn),在浙江省等地區(qū)的數(shù)碼、電腦中始終保持良好的商業(yè)口碑,成績讓我們喜悅,但不會讓我們止步,殘酷的市場磨煉了我們堅強不屈的意志,和諧溫馨的工作環(huán)境,富有營養(yǎng)的公司土壤滋養(yǎng)著我們不斷開拓創(chuàng)新,勇于進(jìn)取的無限潛力,賦悅科技供應(yīng)攜手大家一起走向共同輝煌的未來,回首過去,我們不會因為取得了一點點成績而沾沾自喜,相反的是面對競爭越來越激烈的市場氛圍,我們更要明確自己的不足,做好迎接新挑戰(zhàn)的準(zhǔn)備,要不畏困難,激流勇進(jìn),以一個更嶄新的精神面貌迎接大家,共同走向輝煌回來!