新聞中心 - 賦悅科技(杭州)有限責任公司
-
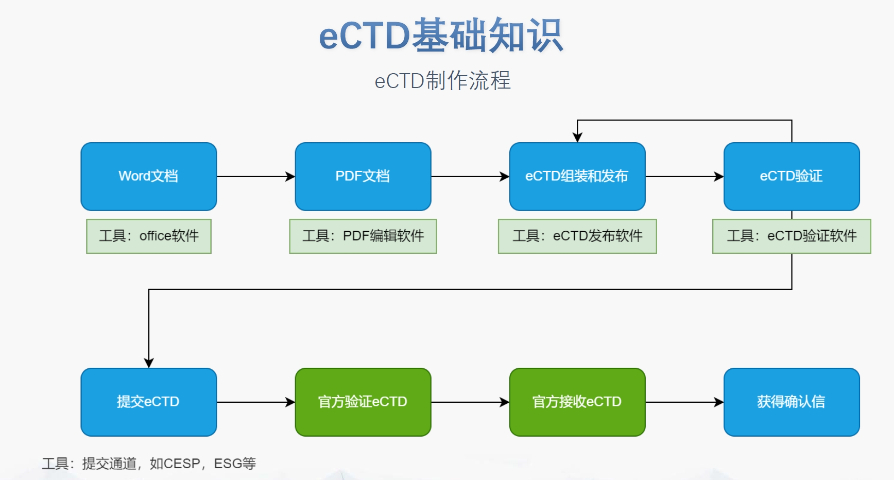 蘇州NDAeCTD軟件
蘇州NDAeCTD軟件eCTD的法規框架與技術規范:歐盟eCTD的法規層級包括指南(Guidelines)、指令(Directive)和法規(Regulation)。其中,法規(如CTR)具有直接法律效力,而指南(如ICH...
發布時間:2025.04.16 -
 楊浦區電子申報eCTD遞交
楊浦區電子申報eCTD遞交澳大利亞的藥品電子通用技術文檔(eCTD)注冊申報體系是澳大利亞y藥品商品管理局(TGA)推動藥品審評現代化的重要舉措。eCTD作為國際通行的電子化注冊申報標準,通過結構化數據格式(如XML)整合了藥...
發布時間:2025.04.16 -
 浦東新區中國eCTD服務介紹
浦東新區中國eCTD服務介紹eCTD 4.0版本的過渡與升級:FDA于2023年啟動eCTD 4.0技術試點,2024年9月正式接收申請,計劃2029年完成全過渡。4.0版本改用HL7 RPS標準替代XML,支持雙向通信和跨申請...
發布時間:2025.04.15 -
 楊浦區仿制藥eCTD名稱
楊浦區仿制藥eCTD名稱技術壁壘與興市場挑戰 非洲和東南亞國家逐步采納eCTD,但其IT基礎設施薄弱導致實施進度滯后。歐盟通過“eCTD全球化倡議”提供技術援助,幫助興市場建立驗證體系和培訓中心。跨國藥企需針對不同區域定制遞...
發布時間:2025.04.14 -
 ANDAeCTD性價比
ANDAeCTD性價比設施費動態調整 API工廠和制劑工廠年費分別約6.8萬和14.5萬美元(2025財年),CMO工廠費用為制劑費的24%。國外工廠需額外支付1.5萬美元跨境檢查費。 ?繳費時限與懲罰 費用需在財年首日(...
發布時間:2025.04.13 -
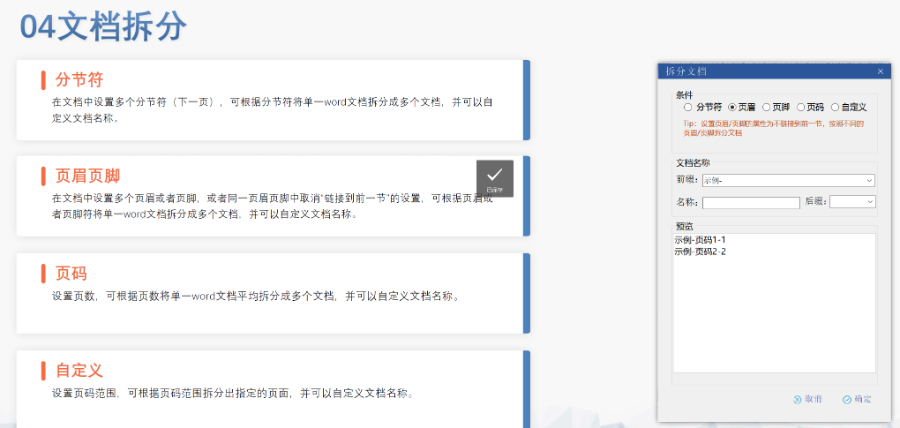 浦東新區CDE eCTD服務放心可靠
浦東新區CDE eCTD服務放心可靠eCTD生命周期管理與變更提交:歐盟要求eCTD申報資料覆蓋藥品全生命周期,包括提交、補充申請及實質性變更。例如,增成員國需提交“附加成員國序列”,審評時間約52-83天;重大變更(如生產工藝調整)需...
發布時間:2025.04.13 -
 浦東新區仿制藥eCTD醫療科技
浦東新區仿制藥eCTD醫療科技從紙質到電子的歷史過渡 2017年前,美國允許紙質與eCTD并行提交,但此后逐步淘汰紙質通道,保留緊急情況下的例外審批。2020年電子化后,所有IND、NDA、ANDA和DMF強制采用eCTD格式。 ...
發布時間:2025.04.12 -
杭州原料藥eCTD文件如何制作
經濟影響與成本效益 盡管初期投入較高(平均每企業需50萬歐元),但eCTD可減少30%的審評延遲成本,長期效益。仿制藥企業通過eCTD復用原研數據,節省80%的申報準備時間。歐盟預算撥款2億歐元資助中...
發布時間:2025.04.12 -
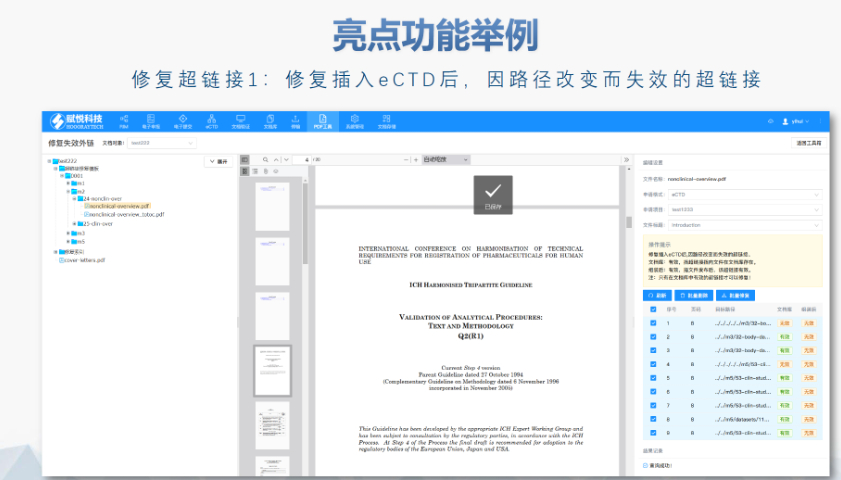 工業園區新藥eCTD性價比高
工業園區新藥eCTD性價比高美國藥物主文件(Drug Master File, DMF)是向FDA提交的機密技術文件,用于支持藥品生產、質量控制及合規性審查。以下為申報的要點和流程總結: DMF概述與類型 ?定義與作用 DMF是...
發布時間:2025.04.11 -
高新區CDE eCTD是什么
DMF維護與合規 ?年度更 即使無變更,每年需提交聲明;重大工藝/設施變更需及時通知客戶并更文件。 ?現場檢查 原料藥企業需通過FDA現場檢查,驗證是否符合ICH Q7 GMP標準,并與DMF內容一致...
發布時間:2025.04.10 -
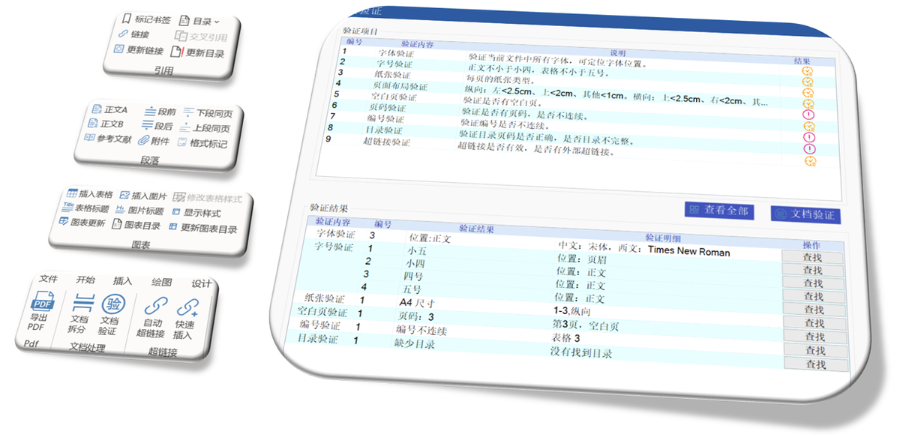 高新區NDAeCTD格式
高新區NDAeCTD格式歐美eCTD實施經驗豐富,中國可借鑒以加速進程。中國可能會經歷從企業自愿eCTD提交到強制eCTD提交的過渡,且將緊隨ICH步伐,尤其在CMC資料整理方面。全球正向eCTD 4.0過渡,中國也不例外,...
發布時間:2025.04.10