新聞中心 - 賦悅科技(杭州)有限責任公司
-
 吳江區NDAeCTD品牌
吳江區NDAeCTD品牌eCTD 4.0版本的過渡與升級:FDA于2023年啟動eCTD 4.0技術試點,2024年9月正式接收申請,計劃2029年完成全過渡。4.0版本改用HL7 RPS標準替代XML,支持雙向通信和跨申請...
發布時間:2025.04.06 -
 高新區國內注冊eCTD服務放心可靠
高新區國內注冊eCTD服務放心可靠歐盟eCTD的歷史沿革與強制實施 歐盟自2003年逐步推進eCTD(電子通用技術文檔)的標準化進程,初要求藥注冊申請(MAA)采用CTD格式。2010年,集中審評程序(CP)率先強制使用eCTD,隨后...
發布時間:2025.04.05 -
 靜安區NDAeCTD服務介紹
靜安區NDAeCTD服務介紹審評效率與時間線優化 eCTD的標準化縮短了審評周期:集中程序平均審評時間從18個月降至12個月,互認程序可在90天內完成成員國意見協調。自動化驗證工具減少了格式錯誤導致的退審率,但復雜藥學數據的科學...
發布時間:2025.04.04 -
 工業園區賦悅科技eCTD便宜
工業園區賦悅科技eCTD便宜區域化差異與多國協作挑戰 歐盟eCTD需兼容成員國特定要求,例如模塊一的行政信息需符合各國語言和法規差異。互認程序(MRP)中,參考成員國(RMS)的評估報告需被其他成員國認可,若出現分歧需由CMDh...
發布時間:2025.04.04 -
 閔行區賦悅科技eCTD遞交
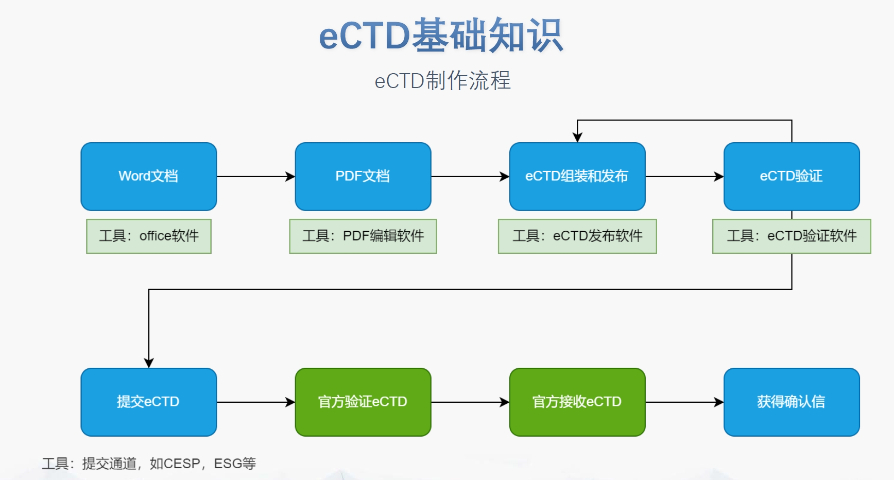
閔行區賦悅科技eCTD遞交eCTD文件制作需遵循嚴格的法規要求和標準化流程,以下是關鍵要點整理:eCTD采用模塊化結構,包含模塊1(行政信息)至模塊5(臨床報告),需按ICH和監機構要求構建目錄樹。顆粒度選擇:文件...
發布時間:2025.04.03 -
 高新區ANDAeCTD性價比
高新區ANDAeCTD性價比內容與格式檢查Word預處理:需檢查拼寫、縮略語、單位格式(如),設置多級列表自動編號(如),統一字體(宋體/TimesNewRoman)和段落格式。重復內容處理:相同劑型不同規格可共用模...
發布時間:2025.04.03 -
 合肥ANDAeCTD哪個品牌好
合肥ANDAeCTD哪個品牌好ANDA一般不需要提供臨床前(動物)和臨床(人體)數據來證明其安全性和有效性(即免毒理和臨床),作為替代,申請人必須合理證明其產品與原研藥相比是生物等效的。 按照《聯邦食品、藥品和化妝品法》第 505...
發布時間:2025.04.02 -
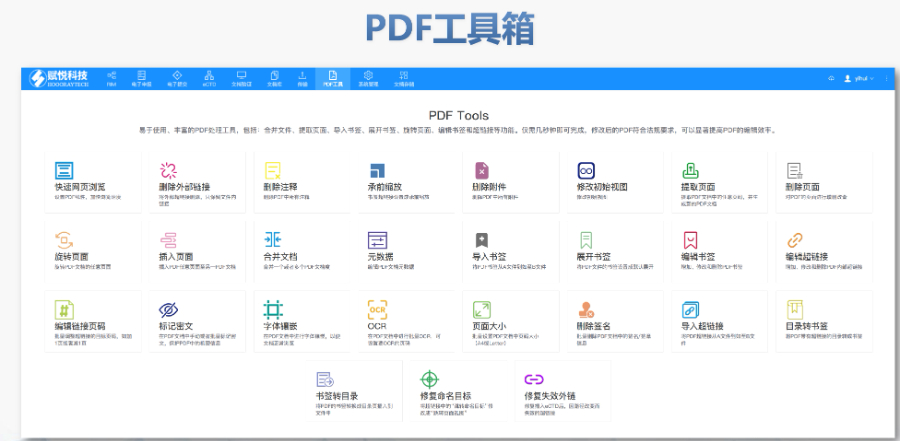 上海原料藥eCTD推薦
上海原料藥eCTD推薦經濟影響與成本效益 盡管初期投入較高(平均每企業需50萬歐元),但eCTD可減少30%的審評延遲成本,長期效益。仿制藥企業通過eCTD復用原研數據,節省80%的申報準備時間。歐盟預算撥款2億歐元資助中...
發布時間:2025.04.01 -
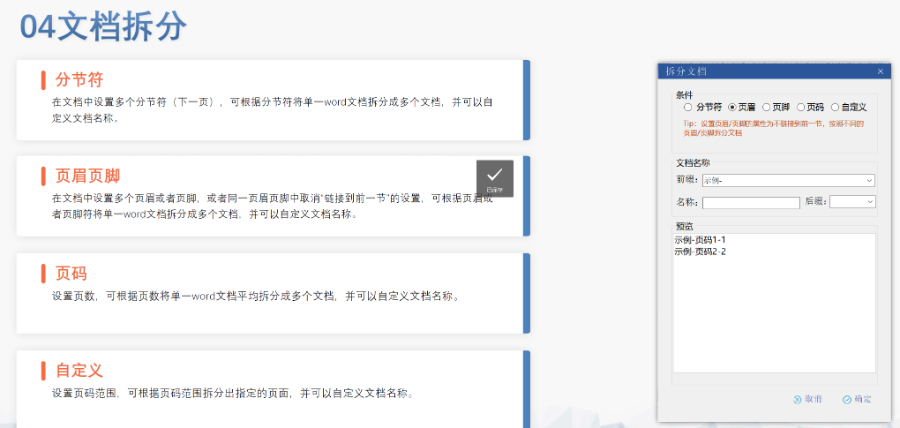 合肥ANDAeCTD格式
合肥ANDAeCTD格式電子遞交的合規性與風險管理 歐盟要求申請人確保電子資料與紙質版本完全一致,若未在規定時間提交紙質文件可能導致注冊終止。驗證過程中,“錯誤”級別問題(如文件命名不規范、XML邏輯錯誤)必須修正,而“警告...
發布時間:2025.03.31 -
合肥國際注冊eCTD注冊系統
多國審評程序與eCTD遞交途徑的適配:歐盟藥品審評程序包括集中(CP)、分散(DCP)、互認(MRP)和國家程序(NP),eCTD需適配不同程序的遞交要求。例如: ?集中審評程序(CP)?:通過EMA...
發布時間:2025.03.31 -
寧波NDAeCTD軟件
eCTD文件制作需遵循嚴格的法規要求和標準化流程,以下是關鍵要點整理:eCTD采用模塊化結構,包含模塊1(行政信息)至模塊5(臨床報告),需按ICH和監機構要求構建目錄樹。顆粒度選擇:文件...
發布時間:2025.03.30